 2023, Vol. 32
2023, Vol. 32
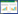



先天性血栓性血小板减少性紫癜(congenital thrombotic thrombocytopenic purpura, CTTP) 是一种超罕见的血栓微血管病变,血小板反应蛋白1型基序成员13(ADAMTS13)的分解素和金属蛋白酶的遗传缺陷引起[1]。该病以微血管病性溶血性贫血、血小板减少、神经精神症状、发热和肾脏受累为临床特征[2]。因其罕见性和临床表现缺乏特异性,该病在新生儿期较难诊断。最终确诊往往需要基因检测证实。本院收治了1例CTTP患儿,回顾性分析以为临床提供经验,现报道如下。本研究经医院伦理委员会批准,伦理批号:IRB-20220348-R。
1 资料与方法患儿胎龄37周,因母“重度子痫前期”剖宫产分娩,过程顺利,人工破膜,羊水清,出生体重3 200 g,APGAR评分10~10分/1~5 min。生后1 h因“新生儿低血糖”入新生儿科。入室时呼吸平稳,无呻吟,无口吐泡沫,未开奶,二便未解。查体:足月儿貌,肤色尚红润,面部三角区及双耳廓瘀紫,呼吸平稳,约58次/min,无明显三凹征,胸廓对称,两肺听诊呼吸音粗,未闻及干湿啰音,心音中,律齐,心率约114次/min,未及明显杂音,腹软,肝脾未及明显肿大,肢体活动好,四肢末梢偏凉,肌张力适于胎龄,胎龄评分37周。辅助检查:末梢血糖1.3 mmol/L。入室诊断:新生儿低血糖。
2 治疗经过患儿入室后予合理保暖,心肺监护,暂禁食,静脉补液、补糖,监测血糖变化;予青霉素预防感染;入室后约7 h因TCB 11.9 mg/dL予光疗,同时完善血常规及生化。血常规示:白细胞计数24.9×109/L,中性粒细胞百分比52.0%,血红蛋白192.0 g/L,红细胞压积0.551,血小板计数32.0×109/L;生化示:总蛋白51.2 g/L,白蛋白36.7 g/L,总胆红素259.5 μmol/L,直接胆红素7.7 μmol/L,间接胆红素251.8 μmol/L。予加强光疗、丙种球蛋白静脉应用。建议换血治疗,家属拒绝。加强光疗及静脉补液等治疗11 h后,复查血清总胆红素310.4 μmol/L,直接胆红素9.8 μmol/L,间接胆红素300.6 μmol/L,直接抗人球蛋白试验阴性,葡萄糖-6-磷酸脱氢酶活性检查正常,血小板计数仍低。生后19 h常规凝血试验检测:凝血酶原时间17.3 s,正常人PT对照12.7 s,国际标准化比率1.47,活化部分凝血活酶时间58.3 s,凝血酶时间18.3 s,纤维蛋白原1.53g/L,无明显异常。生后约20 h予换血治疗,换血完毕血清总胆红素水平203.5 μmol/L,约3.5 h后复查血清总胆红素242.8 μmol/L。予静脉输注白蛋白,继续加强光疗。生后28 h,血清总胆红素308.1 μmol/L,患儿出现易激惹,予再次换血,并予苯巴比妥钠6 mg/kg肌注镇静。第二次换血结束后约1.5 h,血清总胆红素降至162.4 μmol/L。继续间歇光疗,并予输注血小板,静脉补钙等对症支持治疗。行头颅B超、肝胆脾胰B超、泌尿系及肾上腺B超均未见明显异常。生后约45 h复查血清总胆红素126.5 μmol/L,胆红素变化见图 1。患儿略兴奋、易激惹,哭声尖锐,无抽搐、抖动,查体未及明显神经系统异常。予苯巴比妥钠2.5 mg q12h肌注诱导肝细胞酶活性、增加胆红素代谢患儿。高胆红素血症原因未明,完善基因检查。生后约50 h,全血生化显示:乳酸脱氢酶466 U/L。后间歇光疗至生后5 d,复查血常规血小板计数174.0×109/L。生后第9天,完善颅脑MRI提示未见明显异常。生后11 d患儿一般情况好,予出院。生后一个月患儿基因检测结果显示:ADAMTS13基因的1个杂合疑似致病变异c.330+1G > A以及2个杂合的意义不明变异c.2114G > C和c.4013C > A。其中杂合变异c.330+1G > A国外已有报道[3],国内尚无此杂合变异患病的相关报道。

|
| 图 1 总胆红素 |
|
|
CTTP是由染色体9q34上的ADAMTS13基因突变引起的[4]。该突变导致ADAMTS13酶严重缺乏,通常表现为合成和分泌减少或特异性活性降低。ADAMTS13通过调节血管性血友病因子多聚体的大小在止血过程中发挥重要作用[5]。在缺乏ADAMTS13的情况下,超大的血管性血友病因子多聚体进入循环,刺激血小板黏附聚集,从而导致微循环中自发血栓形成。这会导致血小板消耗和受影响器官的缺血,如肾脏、大脑、心脏和肠道。CTTP的发病率非常低,据报道欧洲CTTP的发病率约为0.54/1 000 000[6]。而在亚洲地区,据日本的统计其发病率大约为1/1 100 000[7]。CTTP可分为早发型和晚发型[8],早发型多起病于新生儿及婴儿期,大多数以黄疸起病,治疗过程中逐渐发现血小板减少、溶血性贫血等症状[9]。本案例就是在生后约7 h就出现高胆红素血症,血清总胆红素高达259.5 μmol/L。经过各种对症治疗后,胆红素水平下降不明显,黄疸有延长现象。在国外的确诊病例中,大约有40%~45%患者中出现黄疸延长现象[10]。此外,生化提示:葡萄糖-6-磷酸脱氢酶活性正常,排除葡萄糖-6-磷酸脱氢酶缺乏症,而红细胞形态学检查未见明显异常,不考虑球形红细胞增多症等疾病,其高胆原因有待进一步求证。其次,本案患者在治疗过程中发现血红蛋白逐渐下降,直至出院接近正常水平。网织红细胞增高是溶血的经典参数,在本案例中网织红细胞接近2%,最高达7.24%。结合这几方面,临床故考虑溶血方面的原因,但患儿直接抗人球蛋白试验阴性,排除了免疫性溶血;再者结合乳酸脱氢酶的增高,提示组织有损伤,考虑微血管类病变。此外,该患儿早期即出现重度的血小板减少[11],凝血功能常规检测无明显异常,排除弥散性血管内凝血。该患儿的血小板在使用丙种球蛋白后有所回升但又再次下降,待血浆置换及输注血小板后恢复正常,所以最终临床还是考虑是否存在先天性血栓性血小板减少性紫癜,遂行基因检测以确诊。血小板、血红蛋白以及网织红细胞的变化(表 1)。
| 指标 | 生后8 h | 18 h | 24 h | 28 h | 32 h | 52 h | 70 h | 216 h |
| 血小板(109) | 32 | 15 | 76 | 23 | 166 | 162 | 174 | 385 |
| 血红蛋白(g/L) | 192 | 162 | 125 | 118 | 115 | 108 | 109 | 112 |
| 网织红细胞(%) | / | / | / | 1.92 | 1.98 | 4.51 | 7.24 | 1.65 |
CTTP的临床表现主要是由于血栓形成后各器官缺血引起的一系列表现。比如泌尿系统的急性肾损伤、血尿、肾小管坏死和急性肾功能衰竭;循环系统表现为缺血性心肌损伤、心脏传导异常和充血性心力衰竭;消化系统表现为腹痛、便血和喂养不耐受等[12]。但此案例双肾、输尿管、膀胱超声检查, 肝、胆、胰、脾超声检查及婴儿头颅超声检查均未见明显异常。患儿尿常规,粪常规潜血均阴性。此外,先天性TTP在神经系统方面主要表现为神经状态改变、抽搐、偏瘫、感觉异常、视力障碍和失语症[13]。该患儿在住院期间,神经系统主要表现略显兴奋,易激惹,其他神经系统症状不明显,远期需要进一步评估。由此可见,新生儿期的CTTP临床表现非常不明显,早期诊断需基于多种疾病的鉴别诊断。国外研究指出[14],外周血涂片可见裂体红细胞提供了诊断的方向,它是诊断TTP的重要形态学线索。但本案例是科内第一例,没有做外周血涂片。
CTTP的治疗主要是补充功能性ADAMTS13, 血浆输注是目前主要治疗手段[15]。其他治疗手段包括血浆置换、重组人ADAMTS13、基因治疗等。血浆输注治疗的持续时间取决于其临床疗效,而血小板计数的上升往往是反映治疗效果的最佳指标[16]。但在诊断未明的急性发作期,最常采用的治疗手段仍为血浆置换,可补充功能性ADAMTS13,清除高相对分子质量血管性血友病因子,提高生存率。高伟波等[17]回顾分析了其医院收治的59例血栓性血小板减少性紫癜患者,其中41例进行血浆置换的存活28例,18例未进行血浆置换的存活了6例,差异有统计学意义。此案例系本科首例诊断的CTTP,作为专科医院缺乏ADAMTS13酶活性的检测,早期只能积极的对症处理。在血浆置换及输注血小板后,临床试验室相关指标均有所改善,预后良好。目前,重组人ADAMTS13—BAX930作为一种完全糖基化重组人ADAMTS13,在国际多中心的一期研究中得到其安全性及有效性的验证,且在接受该治疗1个月左右的患者中未发现抗ADAMTS13抗体[18]。CTTP的基因治疗目前还在动物研究阶段,研究[19]探索利用mRNA治疗方法的技术突破,将编码人类ADAMTS13 WT或ADAMTS13变体的mRNA配制成脂质纳米颗粒靶向导入小鼠肝脏,利用肝细胞作为生物反应器持续生产ADAMTS13。这项概念验证研究表明,mRNA治疗可以为TTP治疗提供一种新的方法。同时,mRNA易于大规模生产,性质稳定,免疫原性低,降低产生自身抗体的风险,前景广阔。
利益冲突 所有作者声明无利益冲突
| [1] | Alwan F, Vendramin C, Liesner R, et al. Characterization and treatment of congenital thrombotic throm-bocytopenic purpura[J]. Blood, 2019, 133(15): 1644-1651. DOI:10.1182/blood-2018-11-884700 |
| [2] | 王夕妍, 杨仁池. 血栓性血小板减少性紫癜治疗新进展[J]. 中华血液学杂志, 2019, 40(12): 1055-1059. DOI:10.3760/cma.j.issn.0253-2727.2019.12.020 |
| [3] | Hing ZA, Schiller T, Wu A, et al. Multiple in silico tools predict phenotypic manifestations in congenital thrombotic thrombocytopenic purpura[J]. Br J Haematol, 2013, 160(6): 825-837. DOI:10.1111/bjh.12214 |
| [4] | Lotta LA, Garagiola I, Palla R, et al. ADAMTS13 mutations and polymorphisms in congenital thrombotic thrombocytopenic purpura[J]. Hum Mutat, 2010, 31(1): 11-19. DOI:10.1002/humu.21143 |
| [5] | Furlan M, Robles R, Lammle B. Partial purification and characterization of a protease from human plasma cleaving von willebrand factor to fragments produced by in vivo proteolysis[J]. Blood, 1996, 87(10): 4223-4234. DOI:10.1182/blood.V87.10.4223.bloodjournal87104223 |
| [6] | Reese JA, Muthurajah DS, Kremer Hovinga JA, et al. Children and adults with thrombotic thrombocytopenic purpura associated with severe, acquired Adamts13 deficiency: comparison of incidence, demographic and clinical features[J]. Pediatr Blood Cancer, 2013, 60(10): 1676-1682. DOI:10.1002/pbc.24612 |
| [7] | Kokame K, Kokubo Y, Miyata T. Polymorphisms and mutations of ADAMTS13 in the Japanese population and estimation of the number of patients with Upshaw-Schulman syndrome[J]. J Thromb Haemost, 2011, 9(8): 1654-1656. DOI:10.1111/j.1538-7836.2011.04399.x |
| [8] | Fujimura Y, Kokame K, Yagi H, et al. Hereditary deficiency of ADAMTS13 activity: upshaw-schulman syn-drome[M]//ADAMTS13. Cham: Springer International Publishing, 2015: 73-90. DOI:10.1007/978-3-319-08717-7_5. |
| [9] | 黄抒涵, 党丹, 富鑫, 等. 先天性血栓性血小板减少性紫癜1例报告[J]. 中国实用儿科杂志, 2021, 36(5): 398-400. DOI:10.19538/j.ek2021050619 |
| [10] | Fujimura Y, Matsumoto M, Isonishi A, et al. Natural history of Upshaw-Schulman syndrome based on ADAMTS13 gene analysis in Japan[J]. J Thromb Haemost, 2011, 9(Suppl 1): 283-301. DOI:10.1111/j.1538-7836.2011.04341.x |
| [11] | 中国成人血小板减少症急诊管理共识专家组. 中国成人血小板减少症急诊管理专家共识[J]. 中华急诊医学杂志, 2022, 31(2): 161-168. DOI:10.3760/cma.j.issn.1671-0282.2022.02.005 |
| [12] | Oladapo AO, Ito D, Hibbard C, et al. Patient experience with congenital (hereditary) thrombotic throm-bocytopenic Purpura: a conceptual framework of symptoms and impacts[J]. Patient, 2019, 12(5): 503-512. DOI:10.1007/s40271-019-00365-y |
| [13] | Dong JF, Moake JL, Nolasco L, et al. ADAMTS-13 rapidly cleaves newly secreted ultralarge von Willebrand factor multimers on the endothelial surface under flowing conditions[J]. Blood, 2002, 100(12): 4033-4039. DOI:10.1182/blood-2002-05-1401 |
| [14] | Zini G, d'Onofrio G, Briggs C, et al. ICSH recommendations for identification, diagnostic value, and quantitation of schistocytes[J]. Int J Lab Hematol, 2012, 34(2): 107-116. DOI:10.1111/j.1751-553x.2011.01380.x |
| [15] | 刘婉莹, 肖毅. 遗传性血栓性血小板减少性紫癜的诊断及治疗进展[J]. 中华医学遗传学杂志, 2022, 39(4): 442-446. DOI:10.3760/cma.j.cn511374-20200804-00585 |
| [16] | Matsumoto M, Fujimura Y, Wada H, et al. Diagnostic and treatment guidelines for thrombotic thrombo-cytopenic purpura (TTP)2017 in Japan[J]. Int J Hematol, 2017, 106(1): 3-15. DOI:10.1007/s12185-017-2264-7 |
| [17] | 高伟波, 石茂静, 黄文凤, 等. 59例血栓性血小板减少性紫癜患者临床分析[J]. 中华急诊医学杂志, 2020, 29(1): 106-111. DOI:10.3760/cma.j.issn.1671-0282.2020.01.017 |
| [18] | Scully M, Knöbl P, Kentouche K, et al. Recombinant ADAMTS-13:first-in-human pharmacokinetics and safety in congenital thrombotic thrombocytopenic purpura[J]. Blood, 2017, 130(19): 2055-2063. DOI:10.1182/blood-2017-06-788026 |
| [19] | Liu-Chen SS, Connolly B, Cheng L, et al. mRNA treatment produces sustained expression of enzymatically active human ADAMTS13 in mice[J]. Sci Rep, 2018, 8(1): 7859. DOI:10.1038/s41598-018-26298-4 |